

We are dedicated to providing outstanding customer service and being reachable at all times.
Full-Length Transcript Sequencing (Iso-Seq)
PacBio Iso-seq analysis enables the discovery of new genes, transcripts, and alternative splicing events. The full-length sequences improve genome annotation to identify gene structure, regulatory elements, and coding regions and serve as an ideal method to characterize transcript isoforms within targeted genes or across an entire transcriptome. Based on PacBio’s HiFi Sequencing technology, the Sequel II System, CD Genomics is able to help researchers obtain more accurate information about transcripts and can identify more variable splice sites, new gene locus, new isoforms, fusion genes, etc.
Overview of Pacbio Iso-Seq Analysis
The Iso-Seq method based on Single Molecule, Real-Time (SMRT) Sequencing technology allows generating full-length transcripts. Long-read lengths allow full-length transcripts to be sequenced up to 10 kb or longer, with no assembly required. Since PacBio Iso-seq analysis has the ability to unambiguously determine the full exonic structure of complex genes without the need to assemble, it has been applied to the study of human diseases. Based on the Iso-seq dataset of the MCF-7 breast cancer cell line, scientists discovered additional important information about the well-studied samples, involving new mitochondrial lncRNAs, cancer fusion genes, and novel sample-specific transcripts. Iso-seq analysis is also attractive for research on a variety of important crops and animals because it does not require reference to the genome or pre-existing annotations. This method has been adopted for chicken, rabbit, coffee, cotton, maize, and others. In addition, besides its application to eukaryotes, Iso-seq analysis has been used to study bacteria (E. coli transcriptome) and viruses (transcripts of human cytomegalovirus).
Workflow of Our Pacbio RNA Sequencing Service

Sample Requirements and Preparation
- RNA amount ≥ 5μg.
- RNA concentration ≥ 300ng/μL, OD260/280 =1.8 ~2.0.
- All RNA samples are validated for purity and quantity.
Analysis Pipeline
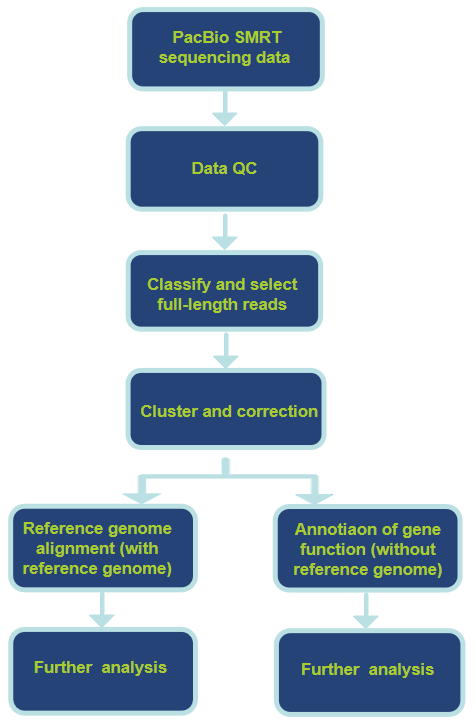
Data Analysis Contents
| Items (without reference genome) | Items (with reference genome) | Bioinformatics tools |
|
-Transcript classification -Transcript clustering and correction -Functional annotation of full-length transcript sequences -LncRNA analysis -CDS prediction |
-Transcript classification -Transcript clustering and correction -Reference genome alignment -Structural annotation of genes -Alternative splicing analysis -Novel RNA prediction -New transcript isoform annotation -Fusion gene analysis -LncRNA analysis -CDS prediction |
-Error correction: IsoCon, IDP, and IDP-de novo -Alignment processing: minimap2 (specialized long read aligners), Cupcake and TAMA -Transcript classification: SQANTI |
Benefits of Our Services
- Advanced experimental platform. Stable running sequencing platform that produces high-quality sequencing data and provides faster project cycle time.
- Professional experimental consultation and design. Senior technical team to guarantee high-quality research results.
- Years of experience in long-read RNA seq analysis. International long-read sequencing pioneer. We have provided accurate and affordable long-read RNA-seq service for years.
With extensive experience in long-read RNA-seq experimental operations and bioinformatics analysis, CD Genomics offers an accurate, rapid, and comprehensive characterization of species and produces reliable results. Furthermore, our end-to-end services guarantee you ultra-fast turnaround time. Thanks for your interest in our methods and services. We are always open to your questions and are happy to support you. Simply complete the contact form and one of our team or business partners will talk to you.
Q&A
What is the output of Iso-Seq?
The Iso-Seq method and bioinformatics workflow yield high-quality, full-length transcript sequences typically spanning 10kb or longer. The accuracy of the HiFi reads enables the identification of SNPs (Single Nucleotide Polymorphisms), UMIs (Unique Molecular Identifiers), and barcodes, which are valuable for single-cell studies.
How does your Iso-Seq Service work?
Iso-Seq (Isoform Sequencing) utilizes the PacBio sequencing technology to capture long, full-length transcripts from RNA samples. The process involves converting RNA molecules into complementary DNA (cDNA), which are then sequenced using PacBio's Single Molecule Real-Time (SMRT) sequencing. The resulting high-fidelity reads are processed bioinformatically to reconstruct complete transcript sequences.
How can I interpret the Iso-Seq results for my research?
Interpreting Iso-Seq results involves understanding the reconstructed transcript isoforms, identifying alternative splicing events, and analyzing gene expression patterns. Our comprehensive bioinformatics analysis package includes detailed reports and visualizations to assist you in drawing meaningful insights from your data.
Can I combine Iso-Seq with other sequencing methods?
Certainly, Our Iso-Seq service can be integrated with other sequencing methods to provide a more comprehensive view of gene expression and transcriptomic diversity. Whether you're interested in combining Iso-Seq with short-read RNA-seq or other techniques, our team can help design a customized approach for your research.
How do I submit samples for Iso-Seq?
Submitting samples for Iso-Seq is a straightforward process. Please follow the Guidelines outlined on our website to prepare your RNA samples. Once ready, complete the submission form, including relevant details about your project and sample specifications. Our team will guide you through the sample submission process and answer any queries you may have.
What types of samples are compatible with Iso-Seq?
We are versatile and compatible with a wide range of sample types, including total RNA, mRNA, and cDNA. Whether you're working with standard cell lines, primary tissues, or challenging samples, our service can be tailored to suit your specific needs. Refer to our Sample Submission Guideline.
Can I use Iso-Seq for single-cell studies?
Absolutely, Iso-Seq is a valuable tool for single-cell studies. With the inclusion of UMIs and barcodes, our service allows for precise identification and differentiation of transcripts from individual cells. This enables in-depth exploration of gene expression profiles and isoform diversity at the single-cell level. Learn more about our Single-Cell Full-Length Transcriptome Sequencing Service.
Can you assist with experimental design for Iso-Seq projects?
Absolutely, we offer expert guidance in experimental design to optimize your Iso-Seq project. Our team of experienced scientists can help you plan the best strategies for sample selection, library preparation, and sequencing depth to ensure the successful attainment of your research objectives. Contact us now for more information.
Reference
- Lee, D. J., & Hong, C. P. (2019). “Transcriptome Atlas by Long-Read RNA Sequencing: Contribution to a Reference Transcriptome.” In Transcriptome Analysis. IntechOpen.
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment