

We are dedicated to providing outstanding customer service and being reachable at all times.

Full-Length 16S/18S/ITS Amplicon Sequencing
CD Genomics is a leading provider of genomic services. We have extensive experience in long-read amplicon sequencing using the PacBio platform. PacBio SMRT long-read sequencing is capable of generating complete, uniform, unbiased coverage spanning long amplicons, allowing for a more realistic restoration of the microbial community structure in the samples.
Introduction of 16S/18S/ITS Amplicon Sequencing
16S/18S/ITS amplicon sequencing, also known as microbial diversity sequencing, refers to the sequencing of PCR products from specific regions such as 16S ribosomal DNA (rDNA), 18S rDNA, Internal Transcribed Spacer (ITS), and functional genes. This method does not require culturing microorganisms, and by detecting sequence variation and abundance in the target region, information on microbial community structure, evolutionary relationships, and microbial relevance to the environment can be obtained from environmental samples.
16S and 18S rDNA are the most commonly used marker genes in metataxonomics. Among them, 16S rRNA gene (~1,500bp) with nine variable regions interspersed between conserved regions, is used to analyze the diversity of bacteria or archaea. 18S rRNA gene (1,500-2,000bp) is utilized to reflect species differences in eukaryotes in a given sample. While ITS (400-900bp) is the spacer DNA between the small-subunit and large-subunit rRNA genes in bacteria, fungi, and archaea. ITS region has been widely utilized for the classification of fungal communities, including ITS1 and ITS2.
Advantages of Full-Length 16S/18S/ITS Amplicon Sequencing Analysis
PacBio SMRT long-read sequencing has overcome the limitations of short reads, allowing researchers to obtain full-length reads of 16s rDNA in bacteria or 18S rDNA and ITS regions in eukaryote.
- High throughput and high-fidelity data: PacBio Sequel's circular-consensus-sequencing (CCS) analysis for generating high accuracy single-molecule consensus sequences of large genomic regions at one time.
- Improved resolution of species identification: High sequence assignment rates for annotation to each taxonomic level.
- Improved accuracy of species identification: Achieving species-level identification.
Sample Requirements
- Sample Type:gDNA/PCR Products, OD260/280=1.8~2.0, no degradation and no contamination.
- DNA amount:
-gDNA ≥100 ng
-PCR fragment ≥500 ng - Sequencing strategy:
PacBio Sequel platform, 2000~20000 CCS reads/per sample
Workflow of Our Service

Analysis Workflow and Contents
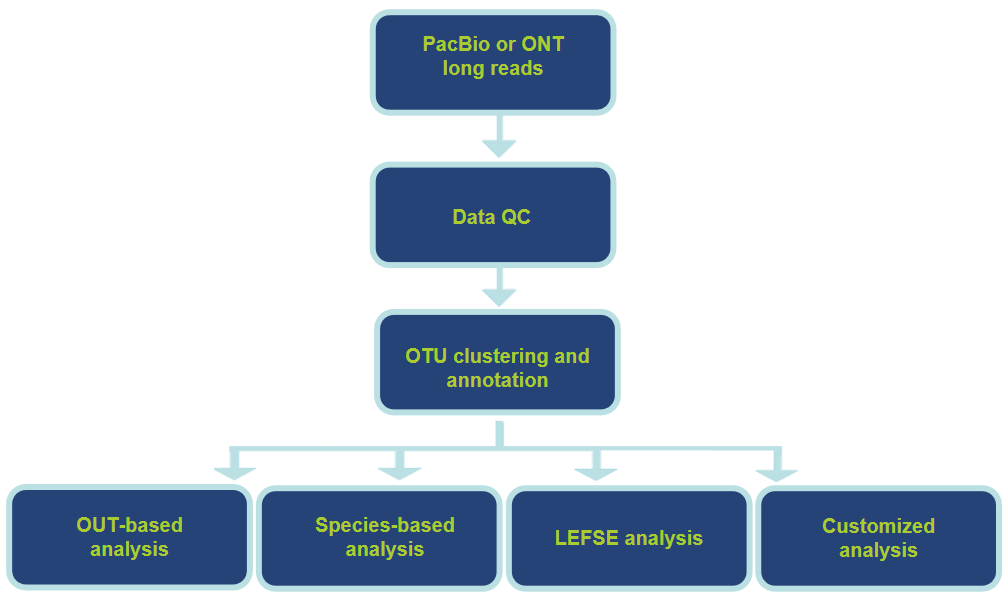
| Standard Analysis | Advanced Analysis |
| -OTU clustering and annotation -Species annotation -Species composition analysis -Alpha diversity analysis -Beta diversity analysis -Functional prediction -Multi statistics |
-Species variation analysis -Correlation analysis of environmental factors -Phylogenetic analysis -Customized analysis |
If you are interested in our full-length 16S/18S/ITS amplicon sequencing service, please feel free to contact us. We've got everything covered for your needs and are ready to assist.
Cases
16s amplicon NGS technology sparked new research on changes in microbial diversity at the phylum/genus level. Full-length amplicon sequencing goes further, exploring relationships between different groups and species within genera.
CD Genomics uses gene sequencing and in-house bioinformatics for multi-omics solutions on microbial data. Our full-length 16s/18s/Its sequencing refines associations from vague classifications to the strain level, aiding key species screening and experimental guidance. If you have any questions, you can contact our technical team.
Long-read 16s rRNA sequencing and metagenome sequencing
16s describes community species, metagenome data validates results, and provides genetic/function annotations, analyzing functional differences in sample communities.
Case: Combining metagenome sequencing and full-length 16s sequencing for functional analysis of the respiratory microbial community revealed significant reductions in the abundance of "nitrate reduction" and "acetyl coenzyme A in butyric acid production" in the disease group. This suggests a close association between the impaired pathways of respiratory microorganisms and the development of chronic obstructive pulmonary disease.
Notably, the disease group exhibited significantly lower abundance in the "nitrate reduction" and "acetyl coenzyme A in butyric acid production," indicating a potential link between the blocked butyric acid production pathway of respiratory microorganisms and the development of LDL.
A high correlation between the predicted functions of the 16s sequencing and the results of metagenomic functional analysis implies that the predicted functions of full-length 16s are more reliable compared to common types of amplicons. This underscores the importance of utilizing advanced sequencing technologies for accurate functional insights into respiratory microbial communities.
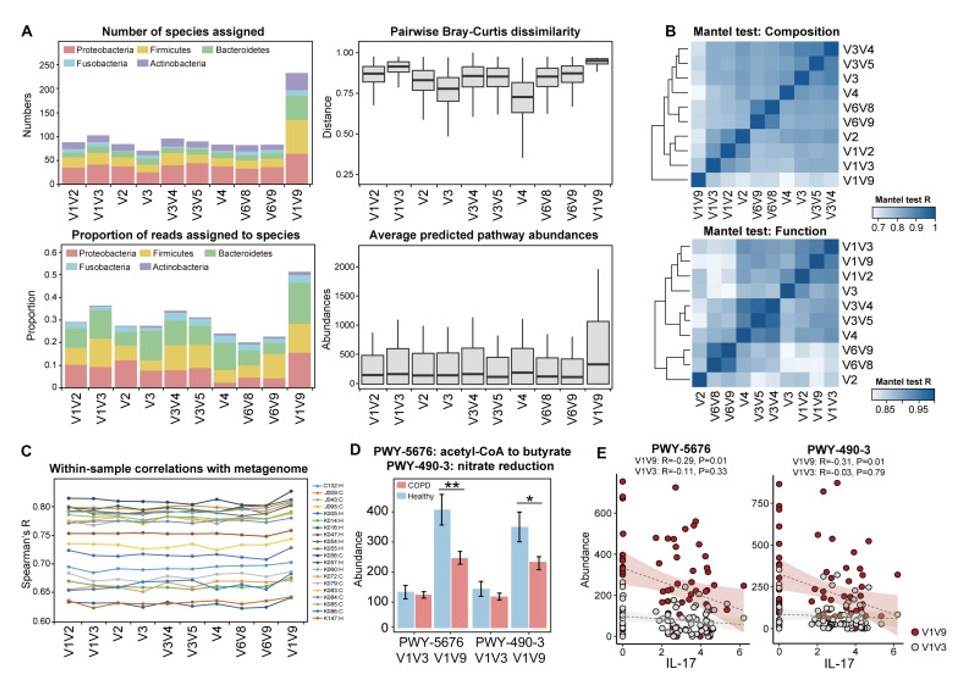 The predicted functions of the 16s sequencing and metagenome. (Wang et al., 2020)
The predicted functions of the 16s sequencing and metagenome. (Wang et al., 2020)
Full-length 16s sequencing and metabolome data analysis
Utilizing full-length 16s sequencing and metabolome data analysis (association and prediction), we establish a connection between the metabolome, the closest biological phenotype to genomics. By associating it with species abundance derived from 16s at diverse molecular levels, we construct a relationship network through statistical modeling. This process allows us to mine potential core regulatory factors, enhancing our understanding of intricate biological interactions.
FAQ
How to choose 16S rRNA and Metagenome sequencing?
When deciding between 16S rRNA and metagenome sequencing, consider the following factors:
16S rRNA Amplicon Sequencing
- Purpose: Ideal for phylogenetic and taxonomic identification of bacteria.
- Analysis: Provides reliable results for community analysis, species composition, and diversity analysis.
- Sequencing Regions: Typically, the V4 region or V3-V4 region is sequenced, or full-length sequencing can be performed.
- Limitation: Restricted to species-level analysis due to sequencing only the 16S region, excluding access to genome gene sequences.
- Advantage: Allows obtaining individual bacterial genomes through assembly and binning techniques.
- Usefulness: Particularly beneficial for isolating difficult-to-culture bacteria.
Consider your specific analysis goals and the nature of the bacteria you are studying to make an informed choice. Or you can contact our technical team for help.
For 16S amplicon sequencing, should I choose NGS or full-length long-read sequencing platforms?
It is recommended to prioritize full-length amplicon sequencing over NGS.
- Resolution: Full-length sequencing provides higher species resolution compared to NGS.
- Analysis Depth: Next-generation sequencing covers only one or two highly variable regions of the 16S gene, limiting the analysis to the genus level.
Choose long-read sequencing for more comprehensive and accurate results tailored to your research needs.
Reference
- Wang, Zhang, et al. "A refined view of airway microbiome in chronic obstructive pulmonary disease at species and strain-levels." Frontiers in Microbiology 11 (2020): 1758.
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment